Harnessing a patient's immune system to attack cancer is the most exciting recent breakthrough in cancer treatment. One of the central mechanisms of action for immunotherapy is reactivation of key killer immune cells, called CD8+ T cells, such that they attack the cancer. However, there are still major gaps in our understanding of how to appropriately mobilise these cells against a tumour.
A recurring theme in cancer immunology is that cancers often evade the immune system by hijacking regulatory processes that benefit the host in other contexts. Two major checkpoints that normally restrain CD8+ T cell immunity are exhaustion and peripheral tolerance. These processes evolved to prevent immunopathology during infection and autoimmunity respectively, but are frequently exploited by cancer to evade destruction by CD8+ T cells. These regulatory processes operate at fundamentally different points in the immune response: peripheral tolerance prevents responses from being initiated within the tumour draining lymph node, while exhaustion dampens existing responses within the tumour.
The Ian Parish laboratory employs a range of non-tumour models, including infection and autoimmune models, to better understand the tolerance and exhaustion processes, and how they can be disrupted. We use the lessons learned from these systems to design and trial new immunotherapies for cancer.
Current projects
Maria Nogueira de Menezes, Sara Roth, Shienny Sampurno, Kelly Ramsbottom, Nicole Saw, Sinead Reading, Christian Deo Deguit, Avraham Travers, Brandon Haugen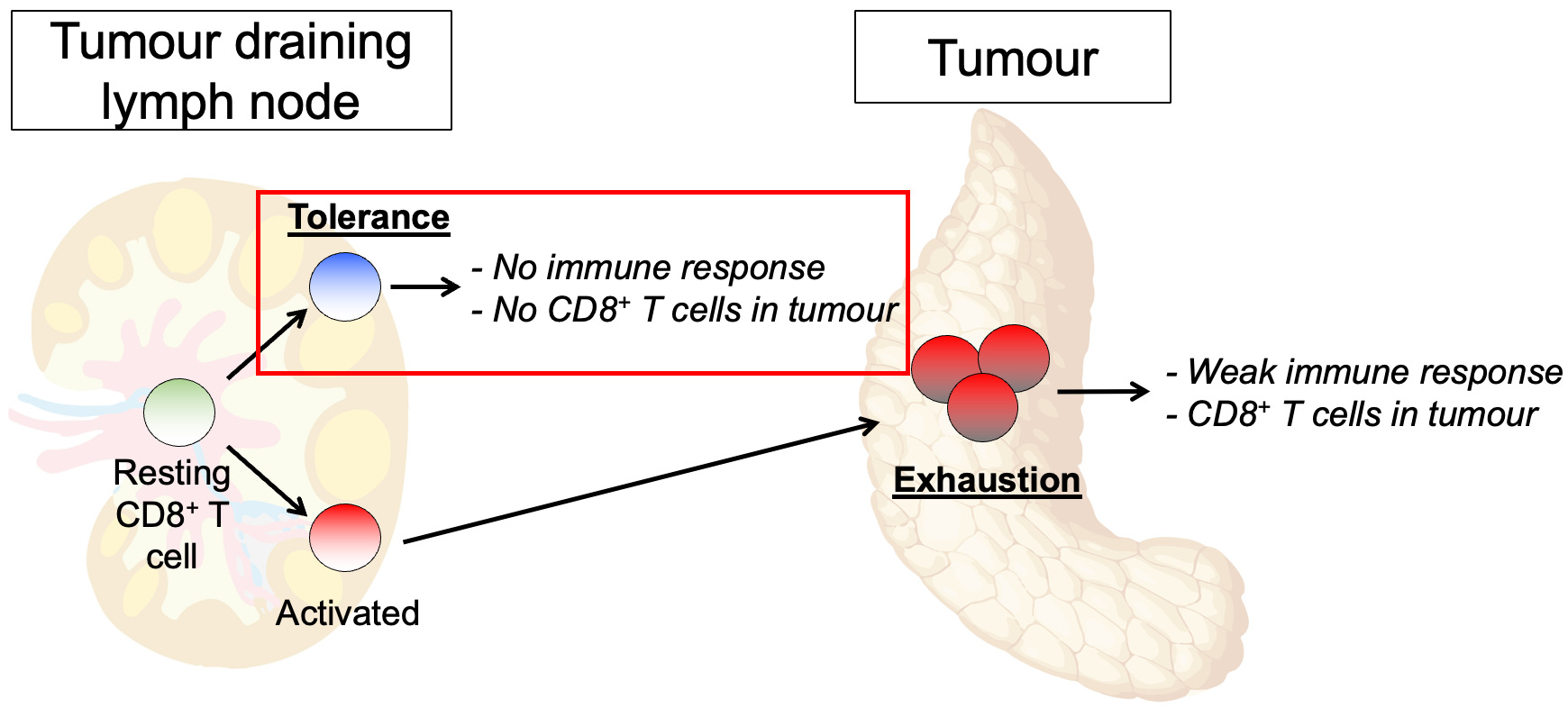 Restraint of anti-tumour T cell immunity by the tolerance and exhaustion regulatory checkpoints
Restraint of anti-tumour T cell immunity by the tolerance and exhaustion regulatory checkpointsLab members
Related links
Related pages